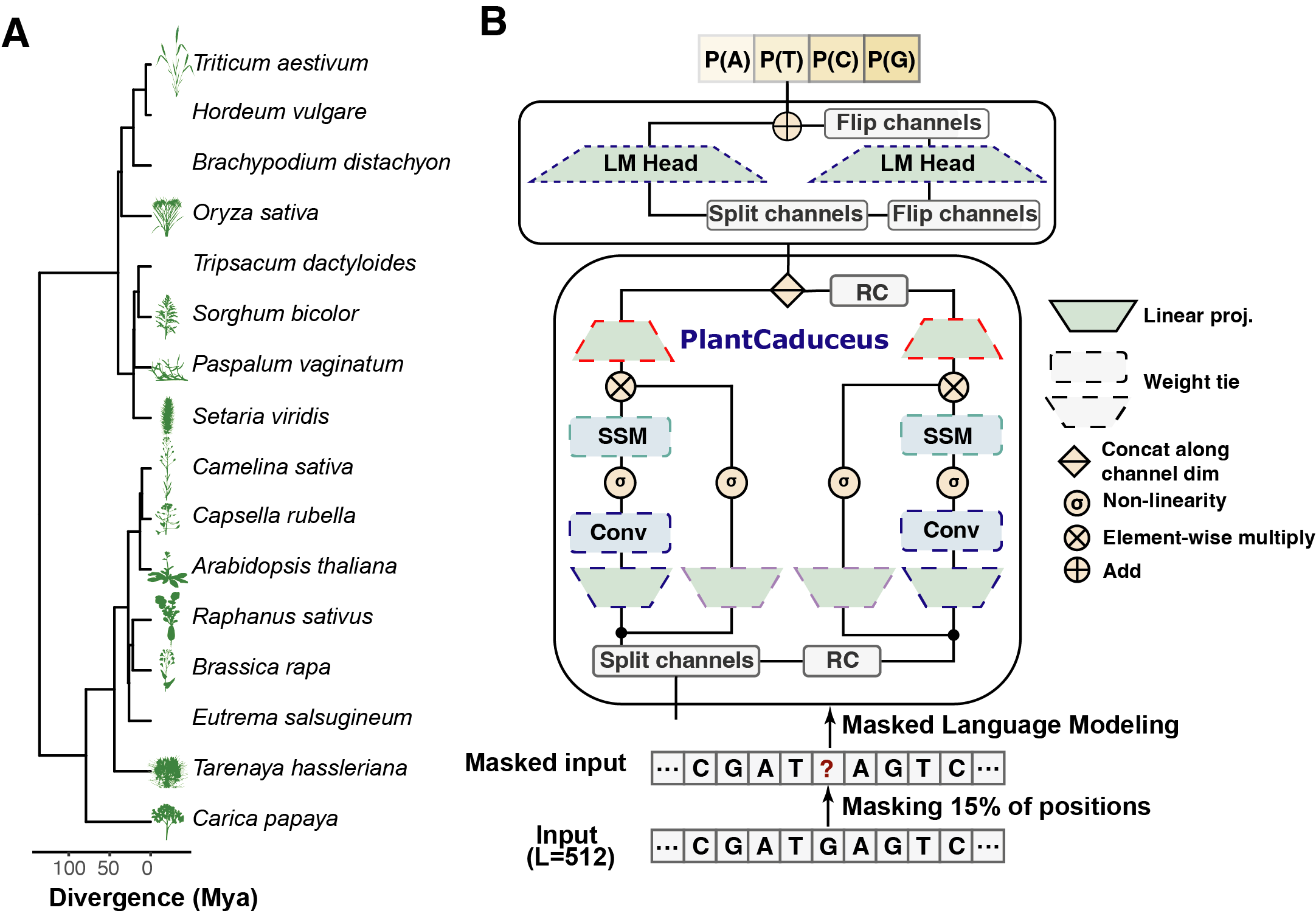
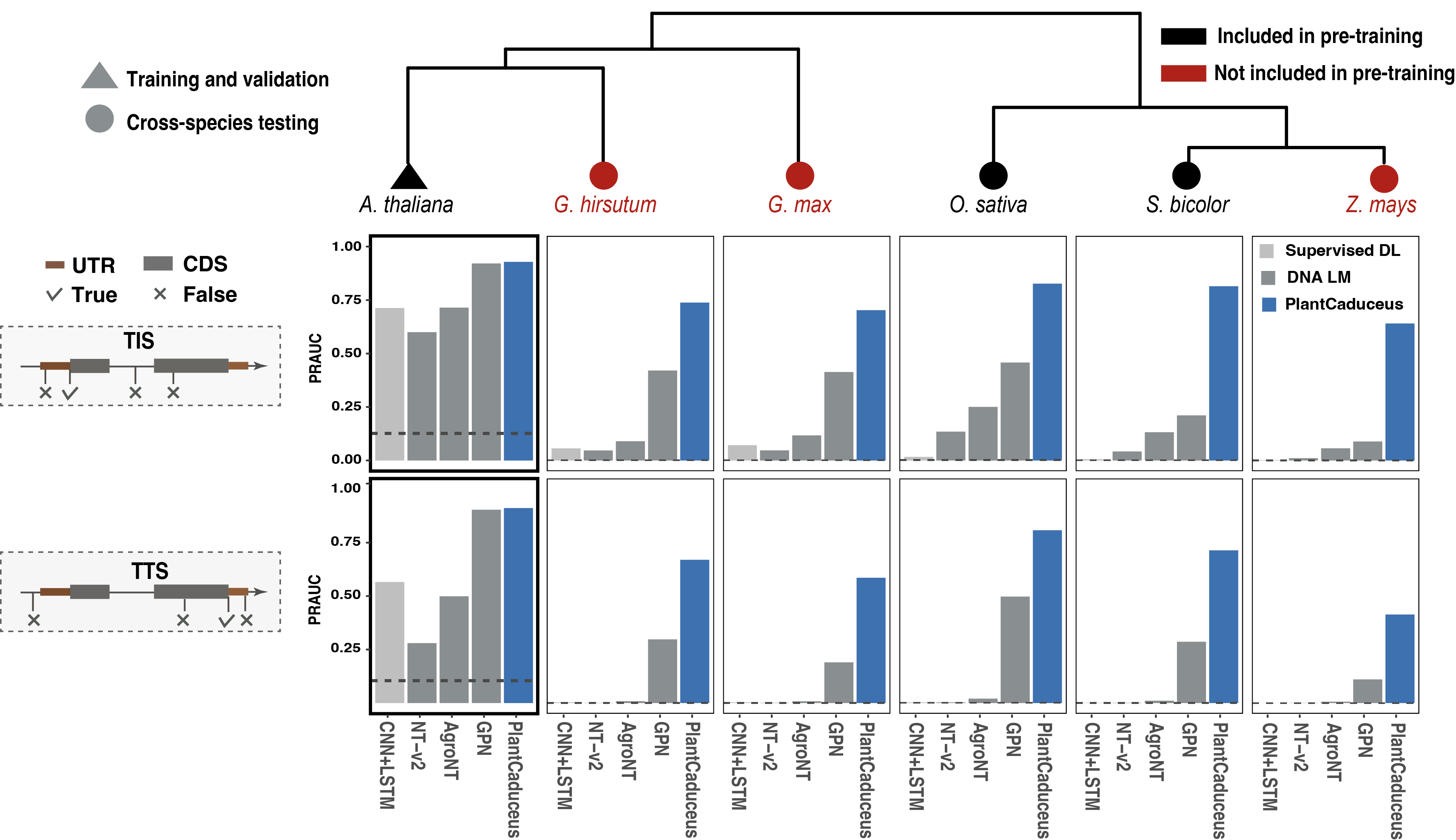
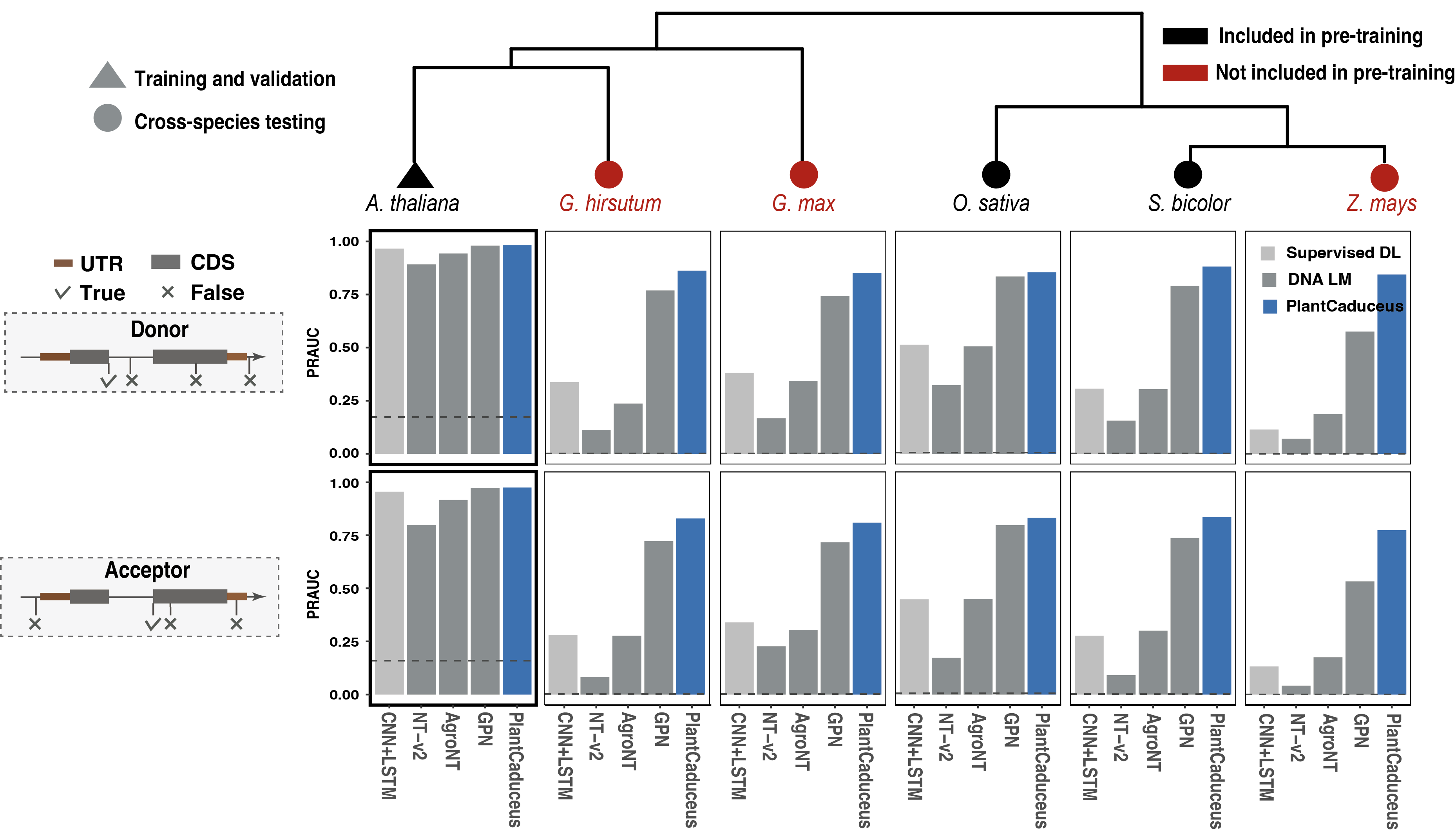
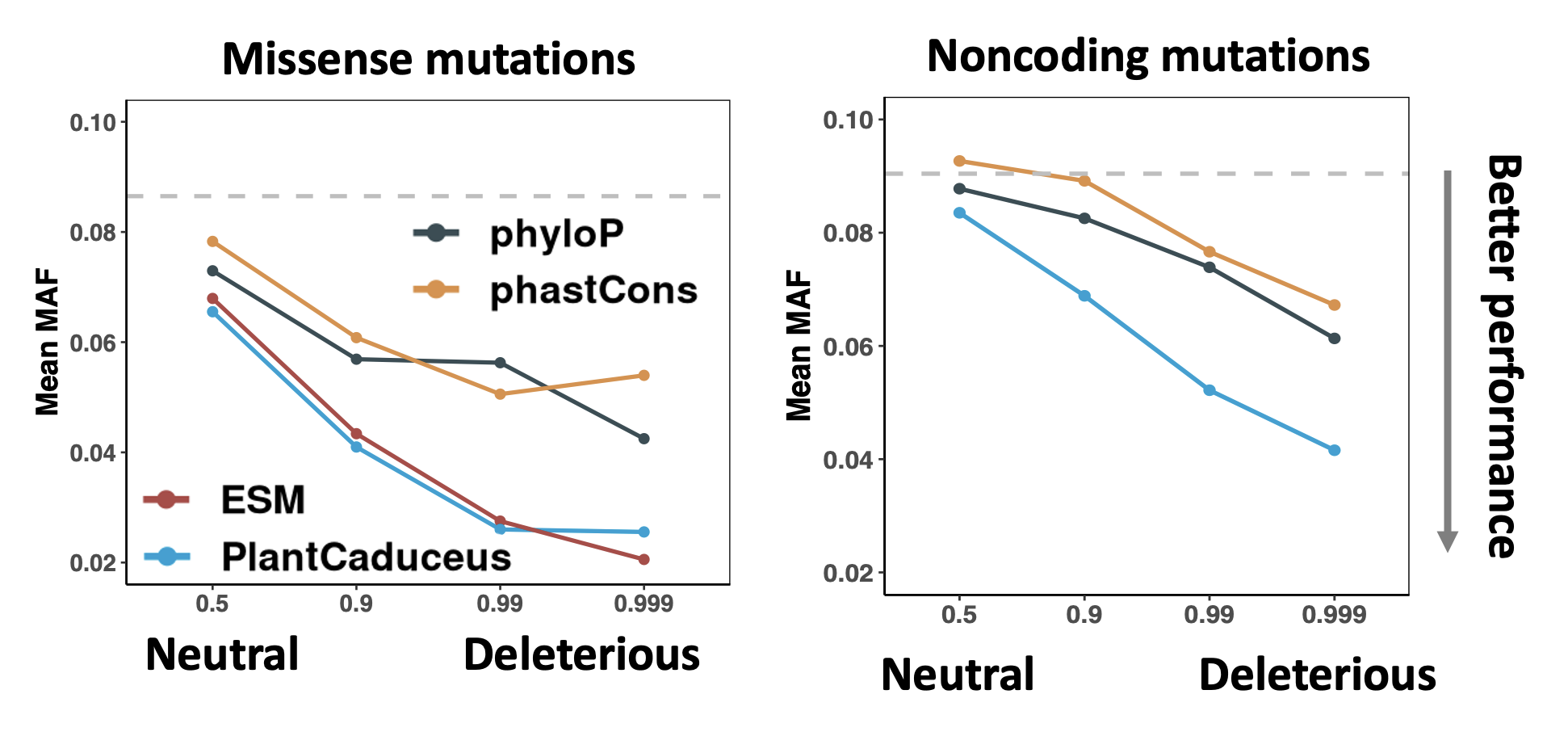
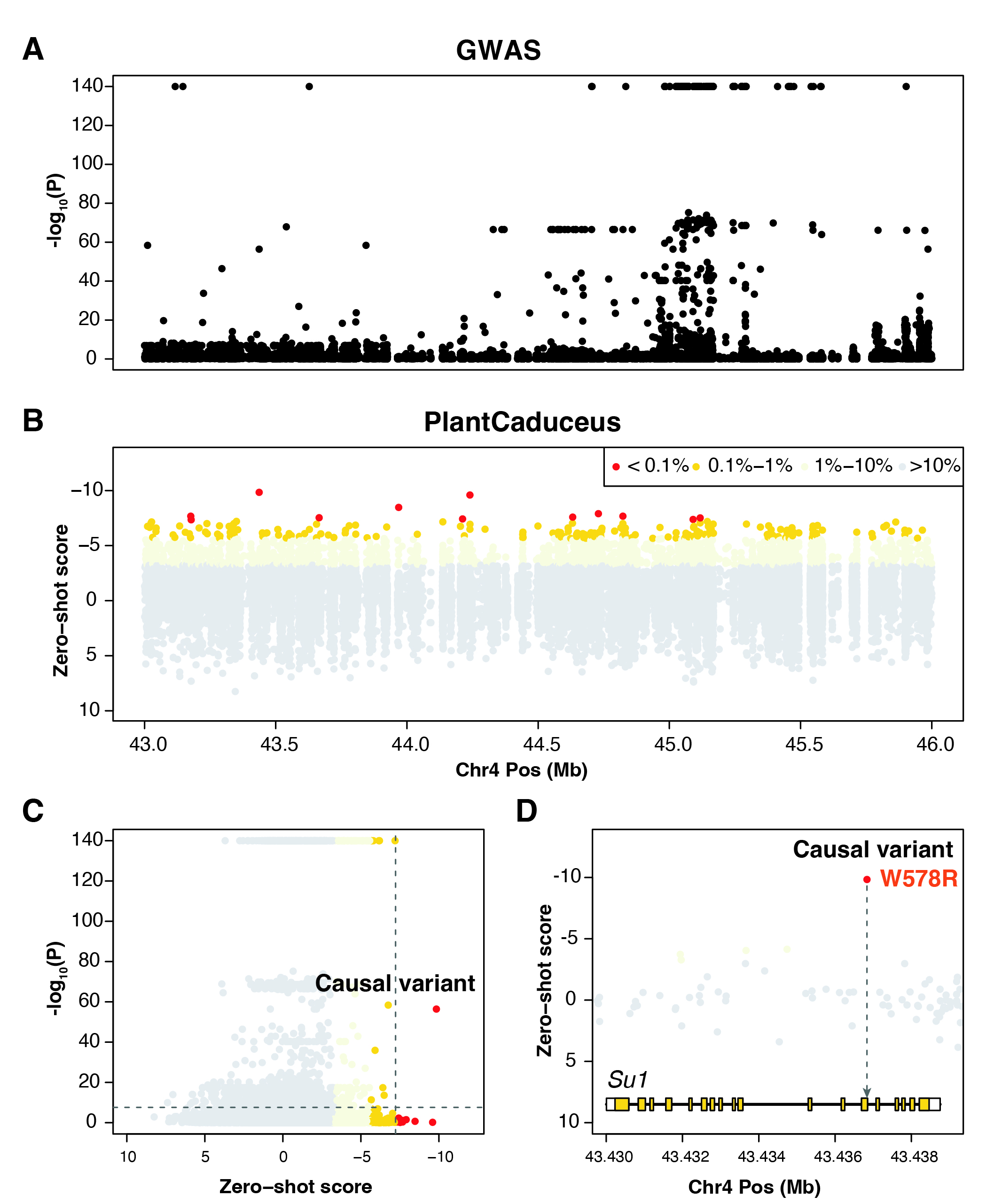
PlantCaduceus (PlantCAD): Cross-species Modeling of Plant Genomes at Single Nucleotide Resolution
PlantCaduceus (PlantCAD): Cross-species Modeling of Plant Genomes at Single Nucleotide Resolution
1
Cornell University
 2
USDA-ARS
2
USDA-ARS
 3
Cold Spring Harbor Laboratory
3
Cold Spring Harbor Laboratory
2
USDA-ARS
3
Cold Spring Harbor Laboratory